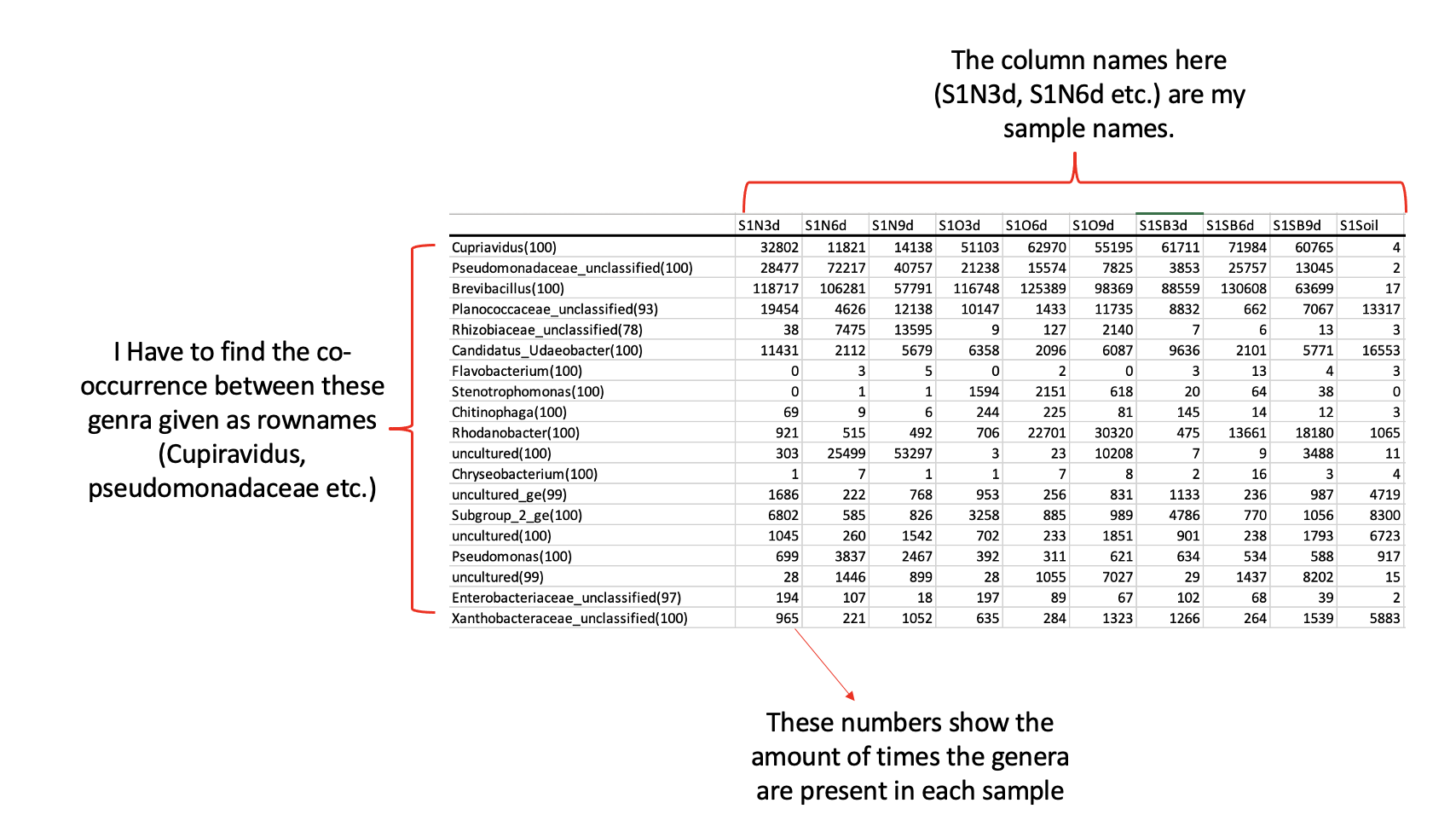
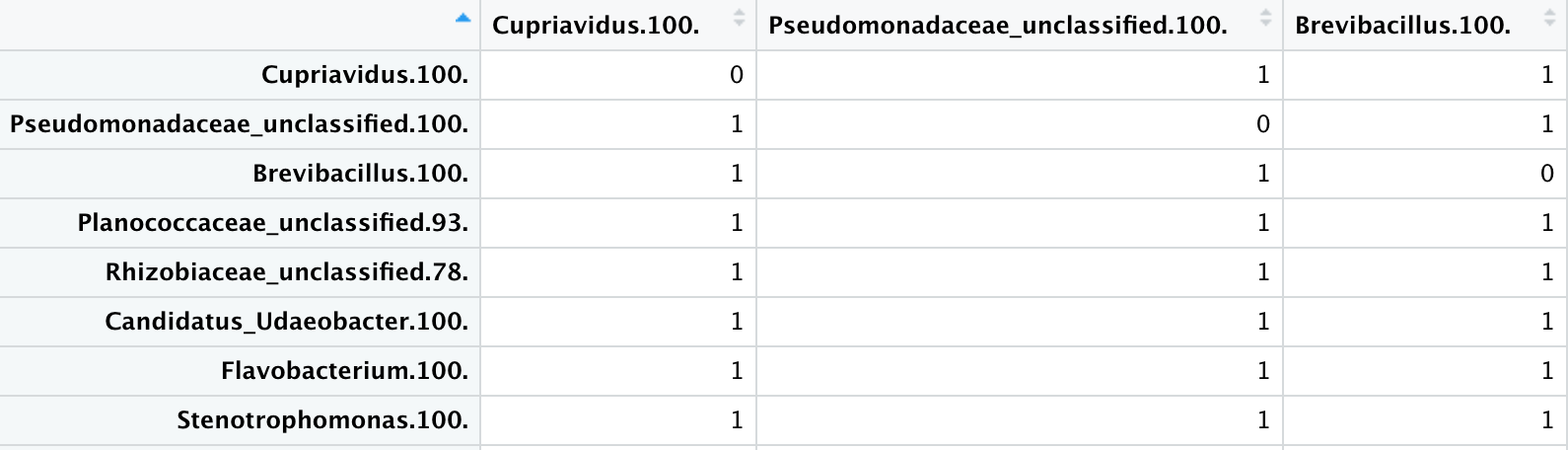
You are right Sir. My diss.mat did not actually have the structure as shown in screen shot.
(Sir, I am giving a smaller part of my data in a copy-paste friendly format as you suggested.)
My matrix that has sample names as column names and the genera as the first column was as follows:
dput(numeric_matrix)
structure(list(S1N3d = c(32802, 28477, 118717, 19454, 38, 11431,
0, 0, 69, 921, 303, 1, 1686, 6802, 1045, 699, 28, 194, 965),
S1N6d = c(11821, 72217, 106281, 4626, 7475, 2112, 3, 1, 9,
515, 25499, 7, 222, 585, 260, 3837, 1446, 107, 221), S1N9d = c(14138,
40757, 57791, 12138, 13595, 5679, 5, 1, 6, 492, 53297, 1,
768, 826, 1542, 2467, 899, 18, 1052), S1O3d = c(51103, 21238,
116748, 10147, 9, 6358, 0, 1594, 244, 706, 3, 1, 953, 3258,
702, 392, 28, 197, 635), S1O6d = c(62970, 15574, 125389,
1433, 127, 2096, 2, 2151, 225, 22701, 23, 7, 256, 885, 233,
311, 1055, 89, 284), S1O9d = c(55195, 7825, 98369, 11735,
2140, 6087, 0, 618, 81, 30320, 10208, 8, 831, 989, 1851,
621, 7027, 67, 1323), S1SB3d = c(61711, 3853, 88559, 8832,
7, 9636, 3, 20, 145, 475, 7, 2, 1133, 4786, 901, 634, 29,
102, 1266), S1SB6d = c(71984, 25757, 130608, 662, 6, 2101,
13, 64, 14, 13661, 9, 16, 236, 770, 238, 534, 1437, 68, 264
), S1SB9d = c(60765, 13045, 63699, 7067, 13, 5771, 4, 38,
12, 18180, 3488, 3, 987, 1056, 1793, 588, 8202, 39, 1539),
S1Soil = c(4, 2, 17, 13317, 3, 16553, 3, 0, 3, 1065, 11,
4, 4719, 8300, 6723, 917, 15, 2, 5883)), class = "data.frame", row.names = c("Cupriavidus.100.",
"Pseudomonadaceae_unclassified.100.", "Brevibacillus.100.", "Planococcaceae_unclassified.93.",
"Rhizobiaceae_unclassified.78.", "Candidatus_Udaeobacter.100.",
"Flavobacterium.100.", "Stenotrophomonas.100.", "Chitinophaga.100.",
"Rhodanobacter.100.", "uncultured.100.", "Chryseobacterium.100.",
"uncultured_ge.99.", "Subgroup_2_ge.100.", "uncultured.100..1",
"Pseudomonas.100.", "uncultured.99.", "Enterobacteriaceae_unclassified.97.",
"Xanthobacteraceae_unclassified.100."))
Then i run the following command:
distances <- vegdist(numeric_matrix, method = "bray")
I get distances as follows:
dput(distances)
structure(c(0.564736701482407, 0.36403669531434, 0.702686441207726,
0.894986835790503, 0.790849593222344, 0.999843796594766, 0.978982622136868,
0.996182385583781, 0.656031231855867, 0.844747458478949, 0.999763337695808,
0.967413029261958, 0.911432057681642, 0.960852572404924, 0.953461762935042,
0.908936675861103, 0.995828766864442, 0.965347250100361, 0.596897763108158,
0.577521718905191, 0.814306902814902, 0.693224848180356, 0.999720252821513,
0.961523290114564, 0.992968943991148, 0.665077521941211, 0.515437214118467,
0.999580410411067, 0.941181361625703, 0.851938895417156, 0.929788184384899,
0.915868944086425, 0.838070635689062, 0.99230930025955, 0.937640651259203,
0.847103573864315, 0.949627309214482, 0.894686047872592, 0.999927169279561,
0.990145662784888, 0.998218274592993, 0.823177728609123, 0.814122955758909,
0.999889652493633, 0.984555033993523, 0.957249032838025, 0.981373159725915,
0.977975921794897, 0.956461098684722, 0.998053052661287, 0.983545198508063,
0.661295469049138, 0.215416414920342, 0.999262108134699, 0.919721399816823,
0.982088030237533, 0.719009005475015, 0.661843859562491, 0.998882194475805,
0.766980889705737, 0.521552163714859, 0.707962826770074, 0.780900498949318,
0.666791388703834, 0.980441668327907, 0.738786305339206, 0.777853283207471,
0.997782137678069, 0.943942652329749, 0.975393253788035, 0.940417433681046,
0.599126104196592, 0.996675616928781, 0.885013066696966, 0.899245209986453,
0.800728663342033, 0.585737947868538, 0.78530943803208, 0.9706947645703,
0.848066223368164, 0.999027366373403, 0.877418373414833, 0.976454132183238,
0.742075736325386, 0.779401513642701, 0.998526681792734, 0.703799535263455,
0.411808786336529, 0.632110886514583, 0.764665584086065, 0.618252074099329,
0.97429665099626, 0.669390568081126, 0.989380530973451, 0.921521997621879,
0.999259001448315, 0.999375545052271, 0.349397590361446, 0.994418132611637,
0.997667020148462, 0.995692187194047, 0.994017946161516, 0.996732511510471,
0.930131004366812, 0.995098403267731, 0.774126534466478, 0.923034975353656,
0.985616684645811, 0.982808022922636, 0.760289961911783, 0.803261666259467,
0.83039190897598, 0.813391877058178, 0.851944996552144, 0.822346368715084,
0.814610190300798, 0.982013267441343, 0.995259246604596, 0.89044289044289,
0.87173585205175, 0.944400481678995, 0.899602385685885, 0.863143631436314,
0.958520072470678, 0.329390892962744, 0.886516853932584, 0.834410943238548,
0.99887748916777, 0.877195592450435, 0.86573793832539, 0.846056516237874,
0.889159902435123, 0.655171150711525, 0.98036010186946, 0.85909747433345,
0.999074253482314, 0.939525415953899, 0.937046364724826, 0.892690685803063,
0.848451583082967, 0.770984125860513, 0.989992638508071, 0.915487391795258,
0.99155476733384, 0.996467304906913, 0.993480245142783, 0.990950226244344,
0.995053423031262, 0.897106109324759, 0.992582702863077, 0.411156612065521,
0.2138557553824, 0.532008248870168, 0.787214068905091, 0.860659618115828,
0.147524085160369, 0.404156619588931, 0.688870774638918, 0.784771699399046,
0.93939601921757, 0.405646573436638, 0.534083992696287, 0.6968466181531,
0.890792158802795, 0.0954038997214485, 0.711287941988064, 0.851384330556257,
0.535690897184021, 0.954867214594518, 0.724388356449789, 0.876632902549773
), Size = 19L, Labels = c("Cupriavidus.100.", "Pseudomonadaceae_unclassified.100.",
"Brevibacillus.100.", "Planococcaceae_unclassified.93.", "Rhizobiaceae_unclassified.78.",
"Candidatus_Udaeobacter.100.", "Flavobacterium.100.", "Stenotrophomonas.100.",
"Chitinophaga.100.", "Rhodanobacter.100.", "uncultured.100.",
"Chryseobacterium.100.", "uncultured_ge.99.", "Subgroup_2_ge.100.",
"uncultured.100..1", "Pseudomonas.100.", "uncultured.99.", "Enterobacteriaceae_unclassified.97.",
"Xanthobacteraceae_unclassified.100."), Diag = FALSE, Upper = FALSE, method = "bray", call = vegdist(x = numeric_matrix,
method = "bray"), class = "dist")
Next, for obtaining an an presence absence matrix I run the following set of commands:
diss.mat <- as.matrix(distances)
diss.cutoff <- 0.6
diss.adj <- ifelse(diss.mat <= diss.cutoff, 1, 0)
Here at this step I do not get the presence absence matrix with the same structure of my numeric_matrix. i.e. sample names as column names and the genra as the first column.
Please tell me what am i missing here to get my desired result. I hope i was able to explain my question more clearly.
Regards
Hira