Hi, I have a communication network between two tissues. Each node in the network has an attribute of correlation with some phenotype(gene expression with BMI). Some node has a quite positive and negative correlation with BMI, and some have a low correlation with BMI. Therefore, in the communication network, some neighbouring nodes (positive nodes) interact with negative nodes. Additionally, negative nodes interact with positive nodes. Biologically, this kind of interaction may not make sense, as positive nodes are only expressed when BMI increase and negative nodes are expressed when BMI is low. Therefore, to make this network meaningful, I want to lower those nodes with a positive correlation with BMI closer to zero; therefore, interaction with a negative node makes sense biologically. The same is true for the other round. However, I don't know how to formulate and code it in R? Additionally, I am wondering if my approach would make sense. Thank you for the help.
Below is the R script for you to understand what I mean.
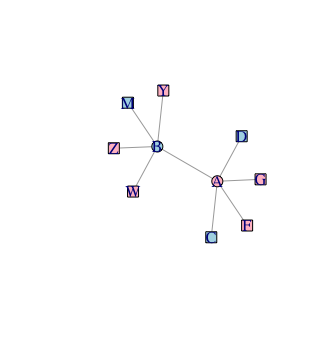
network data
g <- data.frame(source = rep(c("A","B"),c(5,4)),
target= c("D","C","G","F","B",
"W","Z","Y","M"))
g <- graph_from_data_frame(g, directed = F)
node attribuate(corrlation between gene expression and BMI)
node= c(0.3,-0.3,-0.9,-0.6,0.8,0.9,0.5,0.5,0.7,-0.4)
g <- set_vertex_attr(g, "BMI", value = node)
colours represent gene's correlation to BMI
V(g)$color <- ifelse(V(g)$BMI > 0,"red", "lightblue")
Shapes represent tissue
V(g)$shape <- ifelse(names(V(g)) %in% c("A","B"), shapes()[1], shapes()[9])
plot(g)