Hi everyone,
It's the first time I am using TopGo R package and I am an R beginner.
I attached my starting dataset
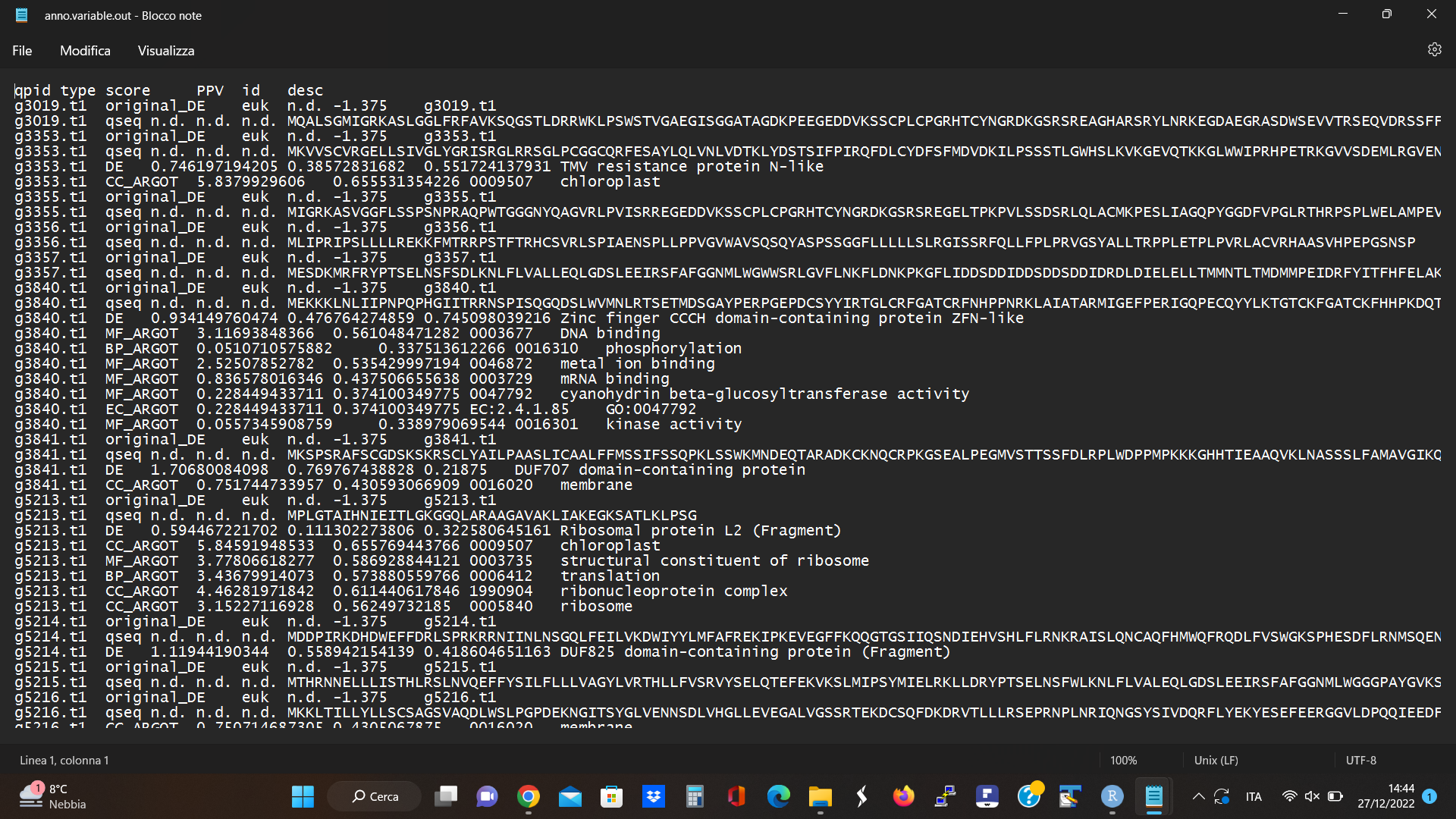
and below I reported the script that I am using. In the last part of the script (#create topGO object), I get this error:
"Error in .local(.Object, ...) : allGenes must be a factor with 2 levels"
Could you please help with this?
Thank you,
Gaia
SCRIPT:
library(tidyverse)
library(data.table)
#if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
#BiocManager::install("topGO")
library(topGO)
#make table from meta data holding grouping information for each sample
#in this case landrace, cultivar ...
getwd()
setwd("C:/Users/iaia-/Desktop/")
background <- fread('anno.variable.out.txt') %>%
as_tibble() %>%
#the ID is the actual GO term
mutate(id = as.numeric(id)) %>%
na.omit() %>%
#according to pannzer2 ARGOT is the best scoring algorithm
filter(str_detect(type ,'ARGOT'),
#choose a PPV value
#according to the pannzer2 manual, there is no 'best' option
# Philipp advised to use 0.4, 0.6, 0.8
# maybe I write a loop to check differences later
PPV >=0.6 ) %>%
#create a ontology column to have the 3 ontology options, which topGO supports
'BP' - biological process, 'MF' - molecular function, 'CC' - cellular component
mutate(ontology=sapply(strsplit(type,'_'),'[',1)) %>%
#select the 3 columns we need
dplyr::select(id,ontology, qpid) %>%
#add GO: to the GO ids
dplyr::mutate(id=paste0('GO:',id))
foreground <- fread('LIST.txt',header=F)
colnames(foreground) <- c("rowname")
#rename type to group to prevent confusion
#dplyr::rename(group=type)
#for all groups together
all_results <- tibble()
for (o in unique(background$ontology)){
filter background for a certain ontology
ont_background <- filter(background, ontology==o)} #BIOLOGICAL PROCESS
annAT <- split(ont_background$qpid,ont_background$id)
filter foreground for a group
fg_genes <- foreground %>% pull(rowname)
ont_background <- ont_background %>%
mutate(present=as.factor(ifelse(qpid %in% fg_genes,1,0))) %>%
dplyr::select(-id) %>% distinct() %>%
pull(present, name = qpid)
#create topGO object
GOdata <-new("topGOdata", ontology = o, allGenes = ont_background, nodeSize = 5,annot=annFUN.GO2genes,GO2genes=annAT)
weight01.fisher <- runTest(GOdata, statistic = "fisher")
results <- GenTable(GOdata, classicFisher=weight01.fisher,topNodes=ifelse(length(GOdata@graph@nodes) < 30,length(GOdata@graph@nodes),30)) %>%
dplyr::rename(pvalue=6) %>%
mutate(ontology=o,
pvalue=as.numeric(pvalue))
all_results <- bind_rows(all_results,results)
}
#all_results %>% pull(GO.ID) %>% writeClipboard()
resultspvalue<- all_results %>% dplyr::select(GO.ID,pvalue)
write_tsv(resultspvalue, "GOTermsrep_pvalue.txt")