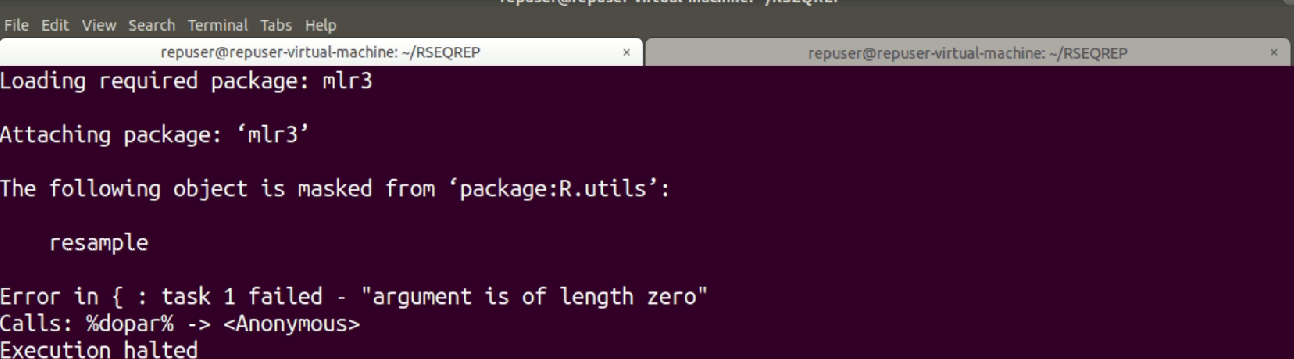
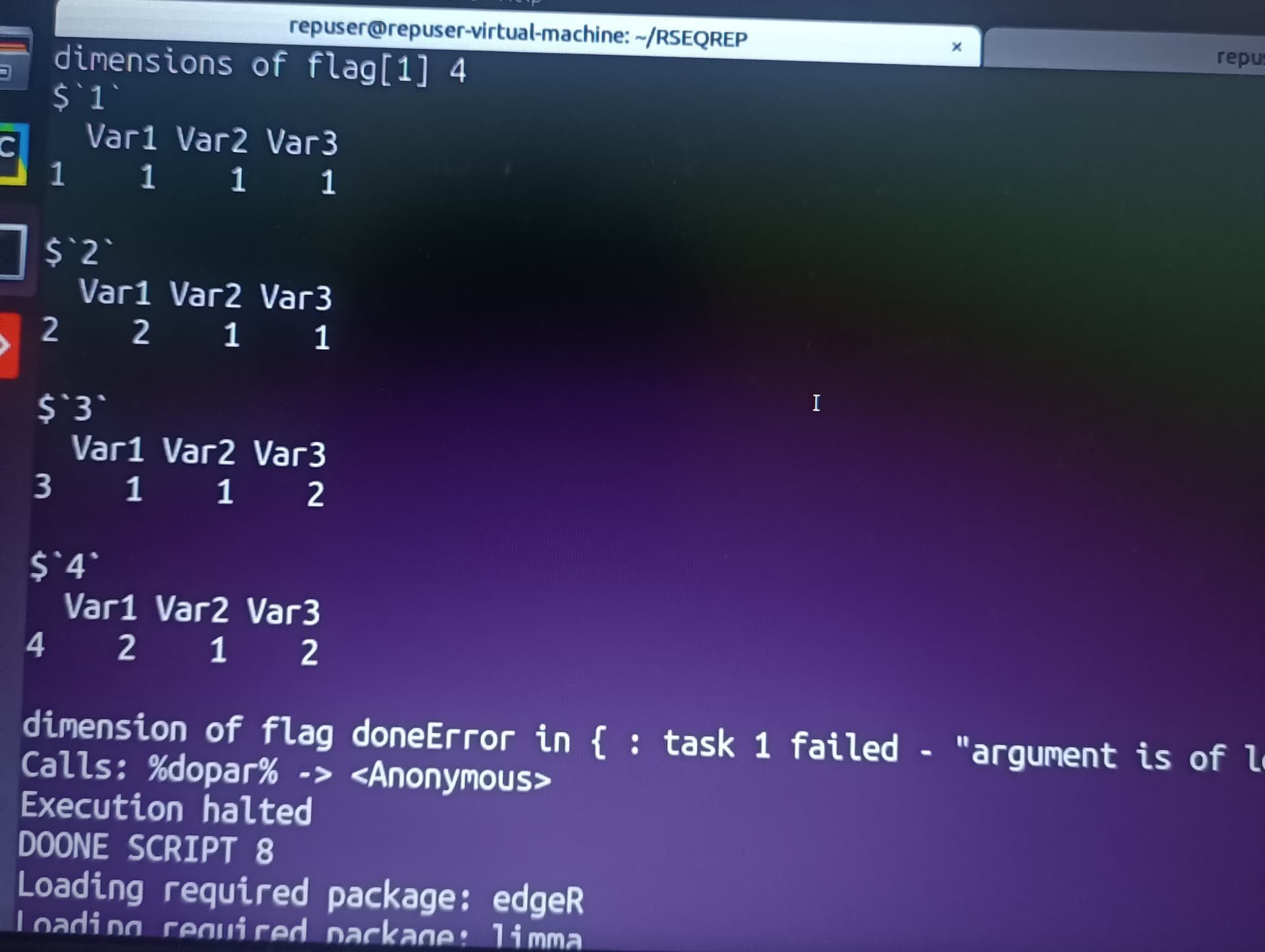
I am running differential expression analysis code using edgeR written by authors of RSEQREP pipeline(opensource) , however the DEG step keeps throwing the following error.
I have managed to pinpoint the error originating from the foreach parallel loop part , but I don't know how to go about it, please help. The code is as shown below. I am using R version 3.6.0 and due to dependency issues its the only one I can work with and ubuntu 18.04
source('init-analysis.r')
## set directories
in.dir.lcpm = paste(res.dir,'lcpm',sep='/');
out.dir.glm = paste(res.dir,'glm',sep='/');
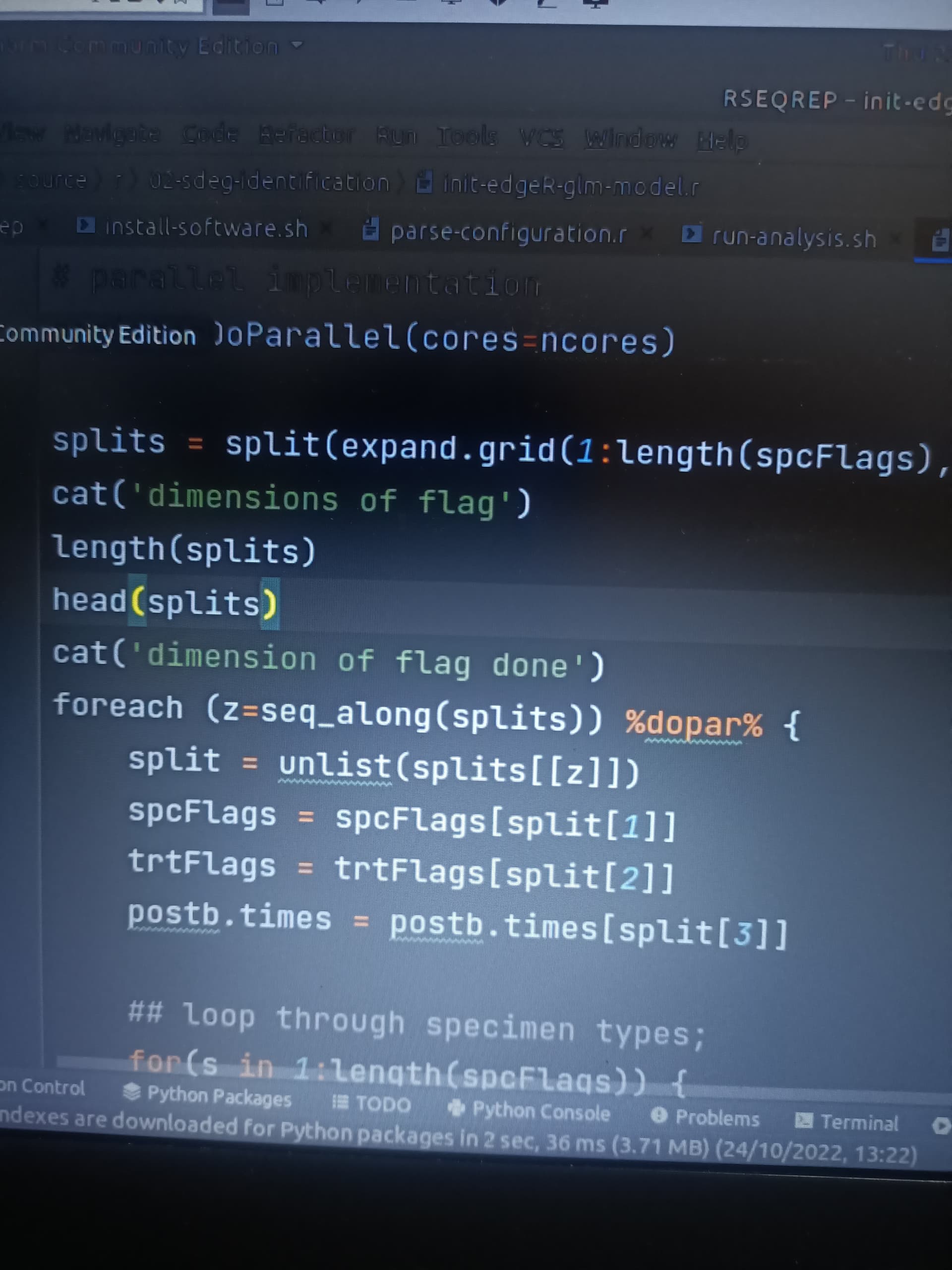
# parallel implementation
registerDoParallel(cores=ncores)
splits = split(expand.grid(1:length(spcFlags),1:length(trtFlags),1:length(postb.times)),1:(length(spcFlags)*length(trtFlags)*length(postb.times)))
foreach (z=1:length(splits)) %dopar% {
split = unlist(splits[[z]])
spcFlags = spcFlags[split[1]]
trtFlags = trtFlags[split[2]]
postb.times = postb.times[split[3]]
## loop through specimen types;
for(s in 1:length(spcFlags)) {
spcFlag = spcFlags[s];
spcLabl = spcFlags[s];
## load edgeR DGE object
load(file=paste(out.dir.glm,'/',spcFlag,'_alltp_edge_r_dge.RData',sep=''));
## loop through treatment groups
for (v in 1:length(trtFlags)) {
trtFlag = trtFlags[v];
## loop through each post treatment time
for(t in 1:length(postb.times)) {
time = postb.times[t];
dge.lab.time=dge.spc;
## select subjects that have both pre post treatment data
subid.base = mta[mta$trt==trtFlag & mta$spct==spcFlag & mta$time %in% b.times & !mta$samid %in% outliers,'subid' ];
subid.post = mta[mta$trt==trtFlag & mta$spct==spcFlag & mta$time==time & !mta$samid %in% outliers,'subid'];
subid.comp = intersect(subid.base,subid.post);
## ensure there are data to compare
if (length(subid.comp)!=0) {
## get ids for treatment time
id.time = mta[mta$trt==trtFlag & mta$time %in% c(b.times,time) & mta$subid %in% subid.comp & !mta$samid %in% outliers,'samid'];
## read gene sets to retain
in.file.gene.sets = paste(in.dir.lcpm,'/',spcFlag,'_posttp_analysis_gene_set.tab.gz',sep='');
gen.lst = read.table(gzfile(in.file.gene.sets),header=F,stringsAsFactors=F);
###########################################
##
## FILTER EDGE_R OBJECT FOR POST treatment TIME
##
###########################################
## retain columns in the count matrix that match selected libraries
id.time.dge = match(intersect(id.time,colnames(dge.lab.time$counts)),colnames(dge.lab.time$counts));
dge.lab.time$counts = dge.lab.time$counts[,id.time.dge];
## retain rows in the count matrix that match genes that passed the low expression cut off
dge.lab.time$counts = dge.lab.time$counts[match(gen.lst[,1],rownames(dge.lab.time$counts)),];
## update sample matrix that match selected libraries
dge.lab.time$samples = dge.lab.time$samples[id.time.dge,]
## align meta data
mta.spc.trt.time = mta[match(colnames(dge.lab.time$counts),mta$samid),];
###########################################
##
## EXPERIMENTAL DESIGN SETUP
##
###########################################
## set all treatment time points to 0;
mta.spc.trt.time[mta.spc.trt.time$time %in% b.times,'time']=0;
## define factors and reference levels
timef = factor(mta.spc.trt.time$time);
subf = factor(mta.spc.trt.time$subid);
## design matrix used for glm
if(glm.model.paired==1) {
design = model.matrix(~0+subf+timef);
} else {
design = model.matrix(~0+timef);
}
## select coefficient of interest (time x treatment interaction)
coefPos = grep(paste('timef',time,sep=''),colnames(design))
###########################################
##
## ESTIMATE DISPERSION
##
###########################################
dge.lab.time=estimateGLMCommonDisp(dge.lab.time,design)
dge.lab.time=estimateGLMTrendedDisp(dge.lab.time,design)
dge.lab.time=estimateGLMTagwiseDisp(dge.lab.time,design)
###########################################
##
## FIT MODEL / LIKELIHOOD RATIO TEST / FILTERING
##
###########################################
dge.lab.time.fit = glmFit(dge.lab.time,design);
## execute likelihood ratio test
dge.lab.time.lrt = glmLRT(dge.lab.time.fit,coef=coefPos);
## get fold change and adjusted p_values
dge.lab.time.all = topTags(dge.lab.time.lrt,adjust.method="BH", sort.by="logFC",n=50000);
## filter gene lists for fold change and adjusted p_value
dge.lab.time.sig = dge.lab.time.all$table[abs(dge.lab.time.all$table$logFC)>=log2(as.numeric(glm.sdeg.fold)) &
dge.lab.time.all$table$FDR<glm.sdeg.qval,];
###########################################
##
## SAVE GLM RESULTS
##
###########################################
## save R objects
out.file.glm = paste(out.dir.glm,'/',spcFlag,'_',trtFlag,'_tp',time,'_glm.RData',sep='')
glm = list();
glm$DGEList = dge.lab.time;
glm$DGEGLM = dge.lab.time.fit;
glm$DGELRT = dge.lab.time.lrt;
save(glm,file=out.file.glm,compress=T);
## save all tabular results
out.file.lab.time.all= paste(out.dir.glm,'/',spcFlag,'_',trtFlag,'_tp',time,'_glm_all.tab',sep='')
write.table(dge.lab.time.all,file=out.file.lab.time.all,sep='\t',quote=F,row.names=T);
R.utils::gzip(out.file.lab.time.all,overwrite=TRUE)
## save significant tabular results if there are anyu
if(nrow(dge.lab.time.sig)>0) {
out.file.lab.time.sig = paste(out.dir.glm,'/',spcFlag,'_',trtFlag,'_tp',time,'_glm_sig.tab',sep='')
write.table(dge.lab.time.sig,file=out.file.lab.time.sig,sep='\t',quote=F,row.names=T);
R.utils::gzip(out.file.lab.time.sig,overwrite=TRUE);
}
}
}
}
}
}