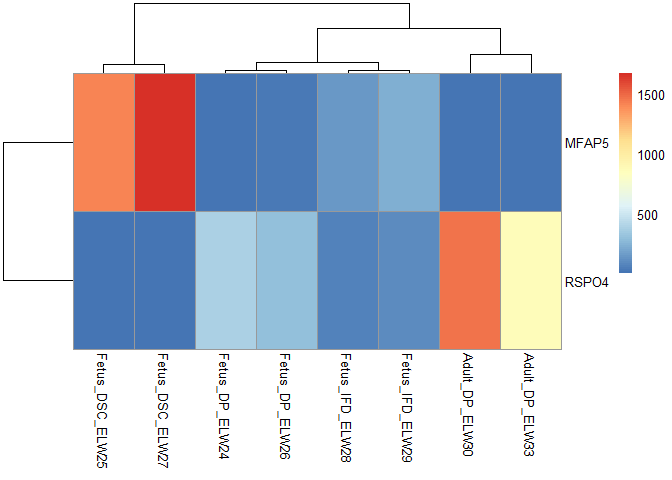
Hello,
I thank you for you answer. Yeah, ok thanks. Then, yes I am using the TPM file (so, I cannot really do edgeR on it? in that case, how can I have comparisons between the conditions and obtaining pvalues for each gene between different conditions?), but let's imagine I could do it.
This is what I did:
charge = read_excel("mypath/GSE149189_CPM_FetusAdult_16623Gene.txt.gz", row.names("refGene"))
head(charge)
group<- factor(c(1,1,2,2,3,3,4,4))
#faire l'analyse
analys <- DGEList(counts=charge[,2:9], genes=charge[1], group = factor(group))
dim(analys)
d <- analys
head(d$counts)
head(cpm(d))
apply(d$counts, 2, sum) # total gene counts per sample
#keep only very expressed genes
keep <- rowSums(cpm(d)>100) >= 2
d <- d[keep,]
dim(d) #3308
#we reset the sample size
d$samples$lib.size <- colSums(d$counts)
d$samples
#normalising
d1 <- calcNormFactors(d)
d1
#we explore the datas
plotMDS(d, method="bcv", col=as.numeric(d$samples$group))
legend("bottomleft", as.character(unique(d$samples$group)), col=1:3, pch=20)
we Estimating the Dispersion
d2 <- estimateCommonDisp(d1, verbose=T)
#Disp = 0.14035 , BCV = 0.3746
d3 <- estimateTagwiseDisp(d2)
plotBCV(d3)
#GLM estimates of dispersion
design.mat <- model.matrix(~ 0 + d$samples$group)
colnames(design.mat) <- levels(d$samples$group)
d4 <- estimateGLMCommonDisp(d,design.mat)
d5 <- estimateGLMTrendedDisp(d4,design.mat, method="power")
You can change method to "auto", "bin.spline", "power", "spline", "bin.loess".
The default is "auto" which chooses "bin.spline" when > 200 tags and "power" otherwise.
d6 <- estimateGLMTagwiseDisp(d5,design.mat)
plotBCV(d6)
#The differential expression
et12 <- exactTest(d3, pair=c(1,2)) # compare groups Adult and DP
e12 <- as.data.frame(et12)
write_xlsx(e12,"mypath_4/7_Adult_VS_DP.xlsx")
et13 <- exactTest(d3, pair=c(1,3)) # compare groups Adult and DSC
e13 <- as.data.frame(et13)
write_xlsx(e13,"/mypaths_4/7_Adult_VS_DSC.xlsx")
et14<- exactTest(d3, pair=c(1,4)) # compare groups Adult and IFD
e14 <- as.data.frame(et14)
write_xlsx(e14,"/mypath_4/7_AdultVS_IFD.xlsx")
et23 <- exactTest(d3, pair=c(2,3)) # compare groups DP and DSC
e23 <- as.data.frame(et23)
write_xlsx(e23,"/mypath_4/7_DP_VS_DSC.xlsx")
et24 <- exactTest(d3, pair=c(2,4)) # compare groups DP and IFD
e24 <- as.data.frame(et24)
write_xlsx(e24,"mypath_4/7_DP_VS_IFD.xlsx")
et34 <- exactTest(d3, pair=c(3,4)) # compare groups DSC and IFD
e34 <- as.data.frame(et34)
write_xlsx(e34,"mypath_4/7_DSC_VS_IFD.xlsx")
###all of these files, e34, e12, e24 etc are the same results
topTags(et12, n=10)
#check the total number of differentially expressed genes
de12 <- decideTestsDGE(et12, adjust.method="BH", p.value=0.05)
summary(de12)
de13 <- decideTestsDGE(et13, adjust.method="BH", p.value=0.05)
summary(de13)
de14 <- decideTestsDGE(et14, adjust.method="BH", p.value=0.05)
summary(de14)
de23 <- decideTestsDGE(et23, adjust.method="BH", p.value=0.05)
summary(de23)
de24 <- decideTestsDGE(et24, adjust.method="BH", p.value=0.05)
summary(de24)
de34 <- decideTestsDGE(et23, adjust.method="BH", p.value=0.05)
summary(de34)
#plot it #you can plot for all of them
de1tags12 <- rownames(d1)[as.logical(de1)]
plotSmear(et12, de.tags=de1tags12)
abline(h = c(-2, 2), col = "purple")